Dossier Preparation, Dossier submission, CTD Format, CTD modules, Regulatory Affairs, NDA, ANDA, MAA

What we learned today is what a common technical document is, the different types of CTD modules, and how to prepare dossiers in CTD format. We have to submit an NDA as per CTD format, and I will explain the criteria for preparing dossiers and what needs to be included in the dossier for regulatory agencies.
What is common technical document (CTD) ?
So, let’s start with the common technical document (CTD). CTD is a document maintained by ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use). It is a set of specifications for the registration of medicines. CTD is used to compile quality data, safety data, and efficacy data in a common format. The CTD format is widely accepted by regulatory authorities in countries like the United States, Europe, Japan, Canada, and Australia.
- CTD is a common technical document which is maintained by ICH
- Basically CTD is a set of specifications which is used for preparing the dossiers to submit to regulator agencies
- These specifications are designed to be used across the US, Europe and japan
- Through CTD we we are compiling quality data, safety data and efficacy data in a common format
- CTD format is widely accepted by the regulatory authorities of other countries as well such as health Canada, Australia.
What are the different types of CTD modules
CTD is divided into 5 Modules which are as follows:
- Module 1 : Administrative and prescribing information.
- Module 2 : CTD summary
- Module 3 : Quality part
- Module 4: Safety data (Non-clinical study reports)
- Module 5 : Efficacy data (Clinical study reports)
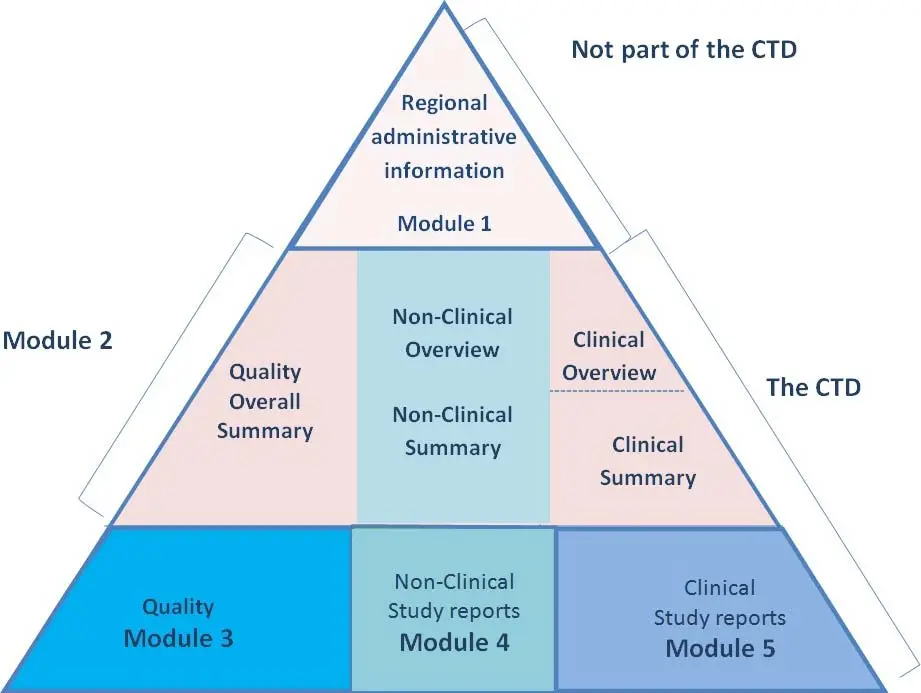
Dossier Preparation As Per CTD Format
we have to submit the ANDA, NDA as per CTD format, Now, let’s talk about the CTD modules. It consists of five modules, each containing specific information about the medicine. When preparing dossiers in the CTD format, it is important to follow the requirements for each module to ensure compliance with regulatory standards.
Module 1 : Administrative and prescribing information.
Module 1 is also called as regional administrative information, it contains regional administrative information, this module 1 is not a part of CTD.
Module 1 which is administrative and prescribing information, we need to submit a cover letter, comprehensive table of contents, application forms, product information, and consultation with the target patient group.
Module 1 : Administrative and prescribing information.
- 1.0 : Cover letters
- 1.1 : Comprehensive table of contents
- 1.2 : Application form
- 1.3 : Product information
- 1.3.1- Summary of product characteristics (SPC’s), labelling and packaging.
- 1.3.2- Mock-up
- 1.3.3- Specimen
- 1.3.4- Consultation with target patient group
- 1.3.5- SPC’s approved in the member states (26 member states + UK)
- 1.3.6- Braille
- 1.4 :Information about the Experts
- 1.5: specific requirements for different types of applications
- 1.6 : Environmental risk assessment report
- 1.7 : Information relating to Orphan market exclusivity
- 1.8: Information relating to pharmacovigilance.
- 1.9 : Information relating to clinical trials.
- 1.10 : Information relating to pediatrics.
- 1.11: Response to Queries.
- 1.12: Additional data
Module 2 : CTD summary
Module 2 is the quality overall summary (QOS) and includes the table of contents, introduction, non-clinical overview, clinical overview, and clinical summary. In the non-clinical overview and clinical summary, we need to submit specific information related to the product.
In module 2 first of all we have to submit
- CTD table of contents (TOC)
- CTD introduction
- Quality overall summary (QOS)
- Non-clinical overview
- Non-clinical summary
- Clinical overview
- Clinical summary
Module 2 : CTD summary
- 2.1: Table of contents
- 2.2: Introduction (General introduction to the pharmaceutical including pharmacology, its mode of action, proposed clinical uses).
- 2.3: Quality overall summary (QOS)
- 2.4: Non-clinical overview
- 2.4.1- General aspects
- 2.4.2- content and structural format
- 2.5: Clinical overview
- 2.5.1- Product development of content rationale
- 2.5.2- Overview of Biopharmaceutics
- 2.5.3- Overview of Clinical pharmacology
- 2.5.4- Overview of Efficacy
- 2.5.5- Overview of safety
- 2.5.6- Benefits and risk conclusions
- 2.5.7- Literature and references
- 2.6: Non-clinical written and tabulated summaries.
- 2.6.1- Pharmacology data
- 2.6.2- Pharmacokinetics data
- 2.6.3- Toxicological data
- 2.7: Clinical summary
- 2.7.1 – Biopharmaceutical studies and associated analytical methods
- 2.7.2 – Clinical pharmacology studies
- 2.7.3 – Clinical efficacy
- 2.7.4 – Clinical safety
- 2.7.5 – Literature references
- 2.7.6 – Synopsis of individual studies
Module 3 : Quality part
Module 3 is the quality part and includes the table of contents, body of data, appendices, regional information and requirements, drug substances, and drug products. We need to provide information about the manufacturing process, control of materials, validation of analytical procedures, and stability data.
Module 3 : Quality
- 3.1: Table of contents
- 3.2: Body of data
- 3.2.S :Drug Substance
- 3.2.S.1 : General Information
- 3.2.S.1.1 – Nomenclature
- 3.2.S.1.2 – Structure
- 3.2.S.1.3 – General properties
- 3.2.S.2 : Manufacture
- 3.2.S.2.1 – Manufacturer details
- 3.2.S.2.2 – Description of the manufacturing process and process controls
- 3.2.S.2.3 – Control of materials
- 3.2.S.2.4 – Control of critical steps and intermediates
- 3.2.S.2.5 – Process validation and/or evaluation
- 3.2.S.2.6 – Manufacturing process development
- 3.2.S.3 : Characterization
- 3.2.S.3.1 – Elucidation of the structures and other characteristics
- 3.2.S.3.2 – Impurities
- 3.2.S.4 : Control of drug substance
- 3.2.S.4.1 – Specification of the drug substance
- 3.2.S.4.2 – Analytical procedures
- 3.2.S.4.3 – Validation of analytical procedures
- 3.2.S.4.4 – Batch analysis (certificate of analysis)
- 3.2.S.4.5 – Justification of specification
- 3.2.S.5 : Reference standards or the materials
- 3.2.S.6 : Container closure system
- 3.2.S.7 : Stability
- 3.2.S.7.1 – Stability summaries and conclusions
- 3.2.S.7.2 – Post approval stability protocol and stability commitments
- 3.2.S.7.3 – Stability data
- 3.2.S.1 : General Information
- 3.2.P :Drug Product
- 3.2.P.1 : Description and composition of the drug product
- 3.2.P.2 : Pharmaceutical development
- 3.2.P.2.1 – Components of the drug product
- 3.2.P.2.2 – Drug product
- 3.2.P.2.3 – Manufacturing process development
- 3.2.P.2.4 – Container closure system
- 3.2.P.2.5 – Microbiological attributes
- 3.2.P.2.6- Compatibility
- 3.2.P.3 : Manufacture
- 3.2.P.3.1 – Manufacturer
- 3.2.P.3.2 – Batch formula
- 3.2.P.3.3 – Description of manufacturing process and the process controls
- 3.2.P.3.4 – Control of critical steps and intermediate
- 3.2.P.3.5 – Process validation and/or evaluation
- 3.2.P.4 : Control of Excipients
- 3.2.P.4.1 – Specifications
- 3.2.P.4.2 – Analytical procedures
- 3.2.P.4.3 – Validation of analytical procedures
- 3.2.P.4.4 – Justification of specification
- 3.2.P.4.5 – Excipients of the human or animal origin
- 3.2.P.4.6 – Novel Excipients
- 3.2.P.5 : Control of the drug product
- 3.2.P.5.1 – Specification of the drug product
- 3.2.P.5.2 – Analytical procedures
- 3.2.P.5.3 – Validation of analytical procedures
- 3.2.P.5.4 – Batch analysis
- 3.2.P.5.5 – Characterization of the impurities
- 3.2.P.5.6 – Justification of the specifications
- 3.2.P.6 : Reference standards or materials
- 3.2.P.7 : Container closure system
- 3.2.P.8 : Stability
- 3.2.P.8.1 – Stability summary and conclusions
- 3.2.P.8.2 – Post approval study stability protocol and stability commitment
- 3.2.P.8.3 – Stability data
- 3.2.R :Regional information/requirements
- 3.2.A : Appendices
- 3.3: List of literature references
Module 4: Safety data (Non-clinical study reports)
Module 4 is the safety part and includes non-clinical study reports. We need to submit information about pharmacology studies, pharmacokinetic studies, and toxicology studies.
Module 4 : Non-clinical study reports
- 4.1: Table of contents
- 4.2: Study reports
- 4.2.1: Pharmacology
- 4.2.1.1 – Primary pharmacodynamic
- 4.2.1.2 – Secondary pharmacodynamics
- 4.2.1.3 – Safety pharmacology
- 4.2.1.4 – Pharmacodynamics drug interactions
- 4.2.2: Pharmacokinetics
- 4.2.2.1 – Analytical methods and validation reports
- 4.2.2.2 – Absorption study
- 4.2.2.3 – Distribution study
- 4.2.2.4 – Metabolism study
- 4.2.2.5 – Excretion study
- 4.2.2.6 – Pharmacokinetic drug interaction
- 4.2.2.7 – Other pharmacokinetic studies
- 4.2.3: Toxicology
- 4.2.3.1 – Single dose toxicity study
- 4.2.3.2 – Repeat those toxicity study
- 4.2.3.3 – Genotoxicity
- 4.2.3.4 – Carcinogenicity
- 4.2.3.5 – Reproductive and development toxicity
- 4.2.3.6 – Local tolerance
- 4.2.3.7 – Other toxicity studies
- 4.2.1: Pharmacology
- 4.3: Literature references
Module 5 : Efficacy data (Clinical study reports)
Module 5 is the clinical study reports and includes the table of contents, table listing of all clinical studies, and individual clinical study reports. We need to provide information about bioavailability studies, bioequivalence studies, and other clinical studies.
Module 5 : Clinical study reports
- 5.1: Table of contents
- 5.2: Tabular listing of all clinical studies
- 5.3: Clinical study reports
- 5.3.1.1: Bioavailability (BA) study reports
- 5.3.1.2: Comparative bioavailability (BA) and bioequivalence (BE) study report
- 5.3.1.3: In-vitro In-vivo correlation (IVIVC) study reports
- 5.3.1.4: Reports of bioanalytical and analytical methods
- 5.3.2.1: Plasma protein binding study reports
- 5.3.2.2: Reports of hepatic metabolism and drug interaction studies
- 5.3.2.3: Reports of studies using human biomaterials
- 5.3.3.1: Healthy subjects pharmacokinetic and initial tolerability study reports
- 5.3.3.2: Patient pharmacokinetic and initial tolerability study reports
- 5.3.3.3: Intrinsic factor pharmacokinetic study reports
- 5.3.3.4: Extrinsic factor pharmacokinetic study reports
- 5.3.3.5: Population pharmacokinetic study reports
- 5.3.4.1: Healthy subject pharmacodynamic pharmacokinetic/pharmacodynamics study reports
- 5.3.4.2: Patient pharmacodynamic pharmacokinetic/pharmacodynamics study reports
- 5.3.5.1: Study reports of control clinical studies
- 5.3.5.2: Study reports of uncontrolled clinical studies
- 5.3.5.3: Reports of analysis of data from more than one study
- 5.3.5.4: Other clinical study reports
- 5.3.6: Reports of post marketing studies
- 5.3.7: Case report forms an individual patient listings
- 5.4: List of key literature references
Overall, preparing dossiers in CTD format requires careful organization and adherence to the specifications outlined in each module. It is important to follow the guidelines set by regulatory agencies to ensure the successful registration of medicines.
Also Read :
21 CFR Part 210 – cGMP in Manufacturing, Processing, Packing, or Holding of Drugs; General
I wants practical experience on ctd and DMF preparation, where should I learn
Very informative