Dissolution method development for generic drug products, Acceptance criteria of dissolution test, ANDA application Submission
In this article you will find the dissolution method development for generic drug products with present and future consideration. Also you will find the biopharmaceutics review approach, how to evaluate the dissolution methods and acceptance criteria for the dissolution methods for genetic drug products mainly the drug product specific dissolution method development and how to set the acceptance criteria and common deficiencies.

Dissolution method development for generic drug products
Following are the steps of Dissolution method development for generic drug products
- Current approaches for dissolution method development for generic drug products
- Present to future considerations.
- Product specific dissolution method development for generic drug products.
- Acceptance criteria for dissolution test.
- Common deficiencies identified in ANDA in the biopharmaceutics section of the application.
- Conclusion
Current approaches for dissolution testing for genetic drug products
Currently ANDA applicants use either the available USP dissolution method for dissolution method development for generic drug products or if USP dissolution method is not available then they use the FDA data-base dissolution method. If these two approaches fail and then they start developing their own new dissolution method for testing genetic drug products.
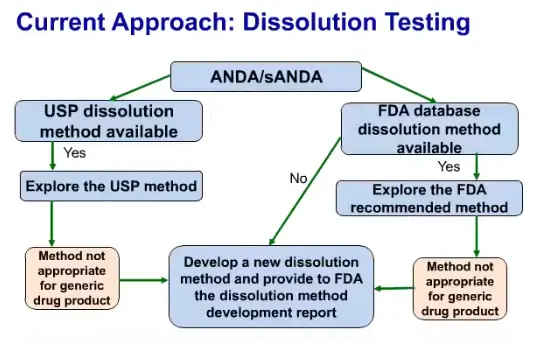
Dissolution method development for generic drug products
Present to future considerations
Considering future Dissolution method development for generic drug products, it is recommended that the applicants to think about product specific dissolution methods making their own methods for their own product and set the specification based on the dissolution data.
Product specific dissolution method development
There are three Critical components of Product specific dissolution method development for generic drug products.
- Evaluation of the dissolution method
- Discriminating dissolution method
- Acceptance criteria of dissolution method
These three are integral to each other for example you may have the right dissolution method with right discriminating ability, however, if we don’t set the specification at the right time point we may lose the disability of the methods.
Evaluation of the dissolution method
- The solubility profile of the API in the physiological pH range.
- Method development report should include supporting in vitro dissolution drug release data.
- The selection of the apparatus or rotation and rotation speed.
- Sink conditions : The applicants should investigate different inventory dissolution media
- Selection of surfactant : if they are using surfactant the use of surfactant and amount and type of surfactant should be justified and whether the sync conditions are maintained or violated should be also clearly specified.
Discriminating dissolution method
Discriminating dissolution method mean by that the method is discriminating if it is able to differentiate drug products batches manufactured under target conditions which is to be marketed formulation from those versus the batches manufactured with variations that could be for genetic drugs or bioequivalent drug products.
Acceptance criteria of dissolution test
Acceptance criteria for Immediate release (IR) Products:
- The acceptance criteria based on the overall data (bio-equivalent (BE) batches and stability batches)
- The acceptance criteria based on collection of complete dissolution profile data (for 12 samples)
- Selection of specification time point should be where NLT 80 % (Q) of the drug is dissolved.
- For slow dissolving products, more than one specification time point needed. (For example, In cases if the drug product even though it’s an IR product if it’s slowly dissolving we may have more than one specification time point and for that you will have an early point and a later on early 15 minutes 20 minutes and then we will have another time point where NLT 80 % (Q) of drug is dissolved).
Acceptance criteria for Extended release (ER) Products:
- The acceptance criteria based on the overall data (bio-equivalent (BE) batches and stability batches)
- The acceptance criteria based on collection of complete dissolution profile data (for 12 samples)
- At least three specification time points covering covering the initial, middle and final phases of the dissolution profile.
- Dissolution acceptance criteria for initial and middle time point should be based on the mean value ± 10 %.
- Dissolution acceptance criteria for last time point should be NLT 80 % (Q) of the drug is dissolved.
The acceptance criteria based on bio-equivalent (BE): when we set the specification based on the available BE data (available BE data could be from the failed BE studies or the adequate acceptable BE studies) and we will use all the available physical chemical properties of the drug product, all the CMC information, in-vitro dissolution, in-vivo pK data to show that the method is discriminatory method.
ANDA application Submission
Dissolution method development for generic drug products. common deficiencies identified in ANDA application Submission are:
- New dissolution method is needed : A new product specific dissolution method should be developed, if USP dissolution method and FDA data-base dissolution method are not available, or the available method are not appropriate for the proposed generic drug product.
- Dissolution method validation data : Generally, Submissions does not include the method validation report and/or method transfer report when method validation is conducted at a different site. It is recommended, to include the validation of the discriminating ability, testing methodology (i.e. robustness etc.) and analytical method used for assay of the dissolution samples (i.e. Linearity, accuracy, precision, etc.).
- Functional scoring : Generally, No dissolution data are submitted to support scoring of the tablets. It is recommended, to submit completed the dissolution profile data for the whole and split tablets using an optimal dissolution method.
- Incomplete stability dissolution data : Generally, stability dissolution data are only provided for the proposed time point. it is hard to set the specification based on the stability data for only proposed time point. so it is recommend that complete dissolution profile data at all time points of the stability program should be included in the ANDA submission.
- SUPAC changes for IR, MR supplemental ANDA : Generally, dissolution data collected on expired lots and pre-change and post-change dissolution data are incomplete. It is recommend that complete pre-change and post-change dissolution profiles should be included in the supplemental ANDA. For MR products dissolution profile data should include more than three time points.
- In vitro release data for semi-solid dosage forms: Generally, In vitro release data is not included to support the post-approval changes for semi-solid products. it is recommend that In vitro release method development report with complete information data supporting the selection of the components of the method and its validation should be submitted.
The importance of communication between the FDA and the applicants through:
- Teleconferences
- Interactive communications
- Advice correspondence
Conclusion
Dissolution method development for generic drug products
Basically the dissolution method is product specific and setting the dissolution specification should be based on the data of the proposed drug product and ANDA submissions should include the complete data in order to optimize regulatory authority review efficiency and approvals and decrease the number of information request deficiencies.
Read more articles
- How to Respond FDA 483 Observations
- Branded vs Generic Drugs: Understanding the Similarities and Differences
- Procedure for WHO GMP Certification in India
- 21 CFR guidelines for pharmaceuticals
Dissolution method development for generic drug products